
A faster, smaller, better way to qPCR
 |
Ultra-Fast Data Acquisition |
|
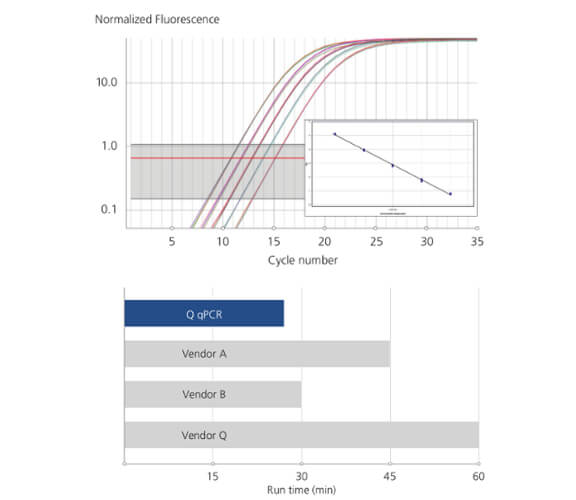
35 cycles in 25 minutes |
|
| Unrivaled Performance | |
|
Detect 2-fold expression level differences |
|
| Portable & Compact | |
|
4.5 lbs – transport without ever calibrating |
|
 |
Scalable & Wireless |
|
Connect up to 10 instruments |
|
 |
Magnetic Induction Technology |
|
Eliminate variability vs block-based cyclers |
Q is intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.
Q – qPCR Instrument
HRM License for Q qPCR Instrument
Q Tubes & Caps (20 racks/box)
Description
Q uses a patented magnetic induction technology to rapidly heat samples coupled with fan forced air for cooling to acquire data in only 25 minutes. Available as a 4 channel model, the robust optical system acquires all channels simultaneously and allows for running the fastest multiplexed assays.
Q's miniature speaker-size and 4.5 pound weight make it the most portable and versatile qPCR machine on the market without ever needing to calibrate. Q also provides scalability as each qPCR machine can process up to 48 samples per run and up to 10 Q's can be connected to a single computer wirelessly via bluetooth enabling up to 480 samples to be processed simultaneously.
A key difference is that Q incorporates a unique spinning aluminum rotor providing superior temperature uniformity of ± 0.05°C versus traditional block-based real time thermal cyclers which rely on multiple peltier heating blocks that can create edge effects resulting in sample variation. Not only does the data give you superior reproducibility, repeatability but enables detection of 2-fold gene expression level differences as well as identification of difficult class IV SNP's requiring melt temperature resolutions of 0.1°C.
Who wouldn't want to take one for a spin?
Q-qPCR Software Tutorials
Details
Specifications
 |
 |
| Front | Back |
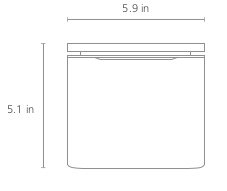
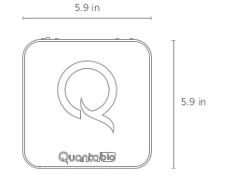
Physical
| Height | 5.1 in |
| Width | 5.9 in |
| Length | 5.9 in |
| Weight | 4.5 lbs |
 |
 |
Thermal Performance
| Temperature Accuracy | ±0.25°C |
| Temperature Uniformity | ±0.05°C |
| Ramp Rates | Heating 4°C/s Cooling 3°C/s |
| Temperature Input Range | 40 – 99°C |
Optical
| Detectors | Photodiode per channel | ||
| Excitation Sources | High power LED for each channel | ||
| Channels | Green | Ex 465nm | Em 510nm |
| Yellow | Ex 540nm | Em 570nm | |
| Orange | Ex 585nm | Em 618nm | |
| Red | Ex 635nm | Em 675nm | |
| Acquisition Time | 1 second | ||
Reaction Tubes
| Samples per Instrument | 48 |
| Reaction Volume Range | 5 – 30µL |
Operating Environment
| Temperature | 18 – 35ºC |
| Relative Humidity | 20 – 80% |
Performance Data
Flexibility from 48 to 480 samples

A qPCR cycler the size of a speaker!

Don’t be fooled by it’s small size. Big things are happening inside.
- Fixed optics & no moving parts
- Never needs optical alignment or calibration
- No reference dyes or cross talk compensation required
- Utilizes proprietary 0.1 ml 4-strip tubes & caps
- supporting volumes of 5 – 30 μl
- High speed centrifugation ensures sample spin down
- Prevents evaporation & condensation with pre-loaded oil in tubes
Q provides ultra-sensitive detection down to single digit copies of DNA.
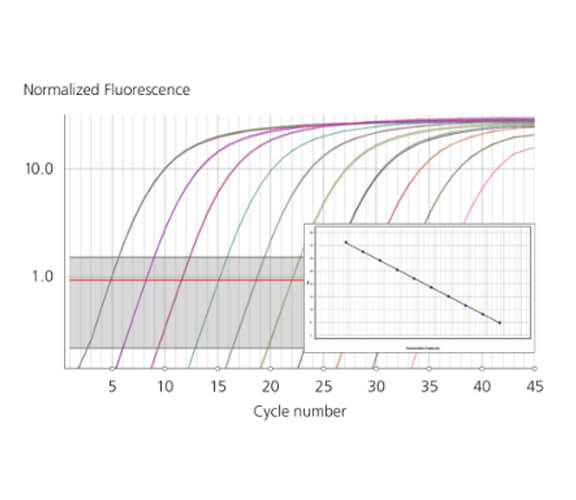
Detect 2-fold Differences
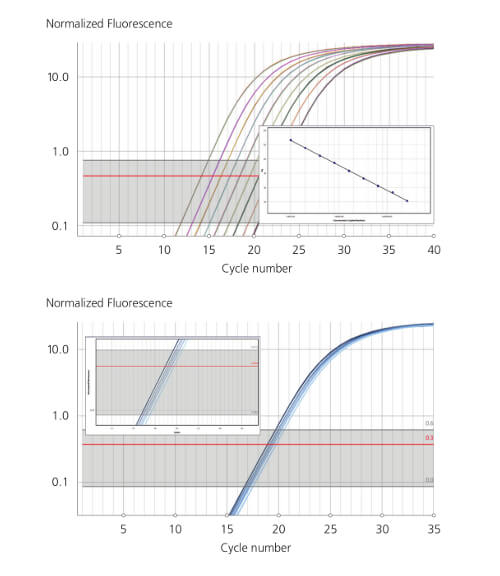
Generate high quality data, fast!
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews

There are no reviews yet