AccuStart II PCR ToughMix
- Stabilized 2X PCR SuperMix enables convenient room-temperature setup and is unaffected by repetitive freeze-thaw
- High-yielding, ultrapure modified Taq DNA polymerase delivers robust, reliable duplex assay performance
- Stringent, ultrapure antibody hotstart ensures sensitive and specific target amplification
- Separate electrophoretic mobility dye reduces risk of post-PCR cross contamination with gel electrophoresis
AccuStart II PCR ToughMix is intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.
AccuStart II PCR ToughMix
Description
AccuStart II PCR ToughMix is a 2X concentrated ready-to-use reaction cocktail for PCR amplification of DNA templates that overcomes many known inhibitors of PCR often present in crude samples extracted from environmental specimens, plant tissues, or animal tissues. The only user supplied components are DNA template and molecular grade water.
A key component to AccuStart PCR ToughMix is an ultra pure, highly processive thermostable DNA polymerase that is combined with high avidity monoclonal antibodies. These antibodies bind the polymerase and keep it inactive prior to the initial PCR denaturation step. This enables specific and efficient primer extension with the convenience of room temperature reaction assembly. Similar to Taq DNA polymerase, the activated polymerase in AccuStart II PCR ToughMix possesses 5'>3' DNA polymerase activity and a double-strand specific 5'>3' exonuclease. The polymerase does not have 3'-exonuclease activity and is free of any contaminating endo or exonuclease activities. PCR products generally contain non-templated dA additions and can be cloned using vectors that have a single 3'-overhanging thymine residue on each end.
GelTrack Loading Dye is a mixture of blue and yellow electrophoresis-tracking dyes that migrate at approximately 4kb and 50 bp. This optional component simplifies post-PCR analysis with gel electrophoresis and eliminates potential for cross contamination by enabling direct transfer of PCR products to the gel sample wells.
The GelTrack Loading Dye solution is not included with the sample size 95142-100.
Details
- 2X concentrated PCR SuperMix containing reaction buffer with optimized concentrations of molecular-grade MgCl2, dNTPs, AccuStart II hot start Taq DNA polymerase, ToughMix additives and stabilizers.
- GelTrack Loading Dye: 50X concentrated mixture blue and yellow electrophoresis-tracking dyes. (Does not contain a density reagent.)
The GelTrack Loading Dye solution is not included with the sample size 95142-100.
Performance Data
Inhibitor Resistance of AccuStart II PCR ToughMix: PCR in the presence of polyphenol spike
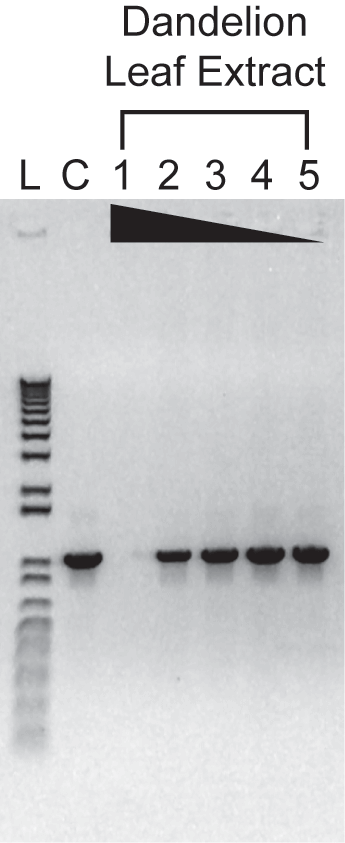
Varying amounts of a polyphenol-rich plant extract (0.2, 0.06, 0.02, 0.006, or 0.002 uL) were added to 25-uL PCRs containing 10,000 copies of a control template. Amplification was carried out for30 cycles of: 94°C, 15s; 60°C, 20s; 72°C, 1 min. 1/5th of each reaction was analyzed on a 01%agarose, 0.5X TBE gel containing 0.25 mg/mL ethidium bromide. As little as 0.002 uL of the crude plant lysate inhibited control reactions with a conventional PCR master mix (data not shown).
Inhibitor Resistance of AccuStart II PCR ToughMix
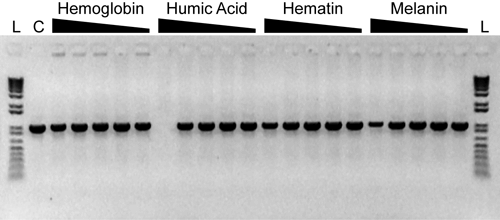
A 1-kb fragment from 1e4 copies of the Tetracyclin resistance gene was amplified in 20-µL reaction volumes according to the recommended protocol. Reactions were challenged with varying concentrations of different PCR inibitors as summarized below. Following a 3 min activation at 94°C; PCR was for 30 cycles of: 94°C, 15s; 60°C, 20s; 72°C, 1 min. 1/5th of each reaction was analyzed on a 01% agarose, 0.5X TBE gel conatining 0.25 mg/mL ethidium bromide.
Hemoglobin: 316 ng/µL, 100 ng/µL, 31.6 ng/µL, 10 ng/µL, 3.16 ng/µL
Humic Acid: 31.6 ng/µL, 10 ng/µL, 3.16 ng/µL, 1 ng/µL, 0.316 ng/µL
Hematin: 100 µM, 31.6 µM, 10 µM, 3.16 µM, 1 µM
Melanin: 10 ng/µL, 3.16 ng/µL, 1 ng/µL, 0.316 ng/µL, 0.1 ng/µL
C: control reactions without inhibitor
L: 1 Kb Plus DNA Ladder (Invitrogen)
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews
AccuStart II PCR ToughMix
We compared the AccuStart II kit to the RepliQa kit, Takara Terra, Takara PrimeStar, and Azura TruFi for air samples that are presumed to be low biomass, low quality, and high amount of impurities/inhibitors (from the air; humans, black carbon, metals, etc.) The AccuStart far outperformed all the other kits. Most kits failed except the Takara Terra (at the temperature/cycle lengths we selected. The kits might have worked at different configurations). But the Terra worked only at diluted concentrations. The AccuStart works for undiluted and diluted samples, showing its superior ability to handle impurities. Bands were very well defined. The only thing was the AccuStart had a lot of non-specific binding so we will try to play around with the temperatures.
I did’t have the time to test it with Eva Green in qPCR.
I got stronger Products by amplifying cDNA directly from Bacteria compare to the enzyme we usually use from NEB OneTAQ Pol