
sparQ HiFi PCR Master Mix
- HiFi DNA polymerase engineered to minimize amplification bias
- Increased amplification efficiency resulting in higher yields
- Uniform coverage across challenging AT- and GC-rich regions
- Robust amplification from input DNA as low as 250 pg
- Cost-effective alternative to KAPA HiFi with improved performance
sparQ HiFi PCR Master Mix is intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.
sparQ HiFi PCR Master Mix
Description
sparQ HiFi PCR Master Mix is a high-fidelity, high-efficiency PCR master mix for NGS workflows requiring DNA library amplification prior to sequencing. The included primer mix allows amplification of DNA libraries flanked by adapters containing the P5 and P7 sequence elements required for Illumina® sequencing platforms. The hot-start, proofreading DNA polymerase used in the sparQ HiFi PCR Master Mix is specifically engineered to improve library amplification efficiency while reducing PCR-derived artifacts, resulting in higher library yields and better coverage uniformity. This kit supports low DNA input from 250 pg and efficient amplification of AT- and GC rich regions with minimal bias.


Details
Performance Data
sparQ HiFi PCR Master Mix fragment size distribution and the quality of the amplified DNA
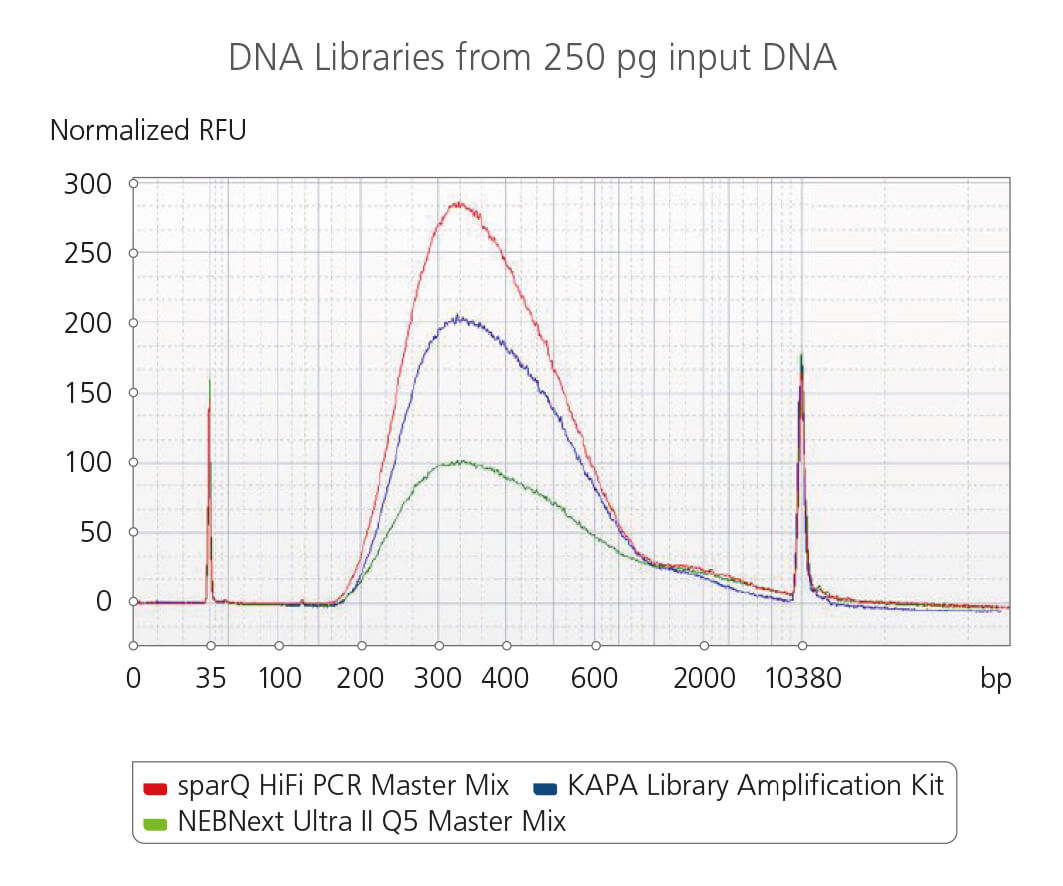
The fragment size distribution and the quality of the amplified DNA libraries from 250 pg input DNA were analyzed using a high sensitivity DNA analysis kit on a Bioanalyzer. Libraries were amplified using sparQ HiFi PCR Master Mix (red), KAPA Library Amplification kit (blue), or NEBNext Ultra II Q5 Master Mix (green) with identical amplification cycle numbers (16 cycles for 250 pg input DNA).
sparQ HiFi PCR Master Mix coverage
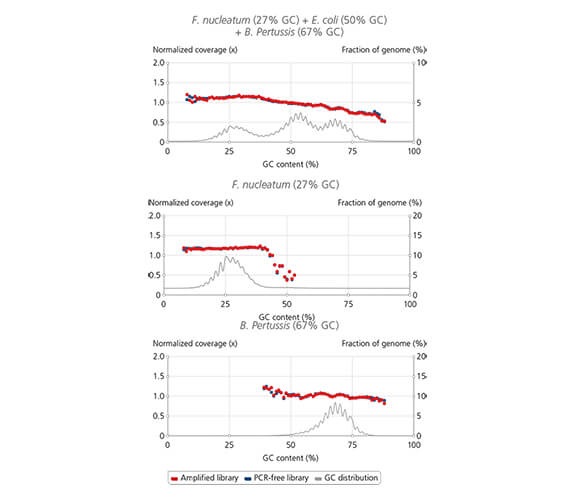
Library amplification with sparQ HiFi PCR Master Mix resulted in uniform coverage across the wide range of GC-content. Libraries were prepared by using sparQ DNA Library Prep Kit with 100 ng input DNA.
Coverage depth against GC-content of libraries amplified by sparQ HiFi PCR Master Mix (red) were compared to corresponding libraries without amplification (dark blue: PCR-free library). GC content distribution of targeted genomes is indicated by gray line.
sparQ HiFi PCR Master Mix Library Yield Analysis
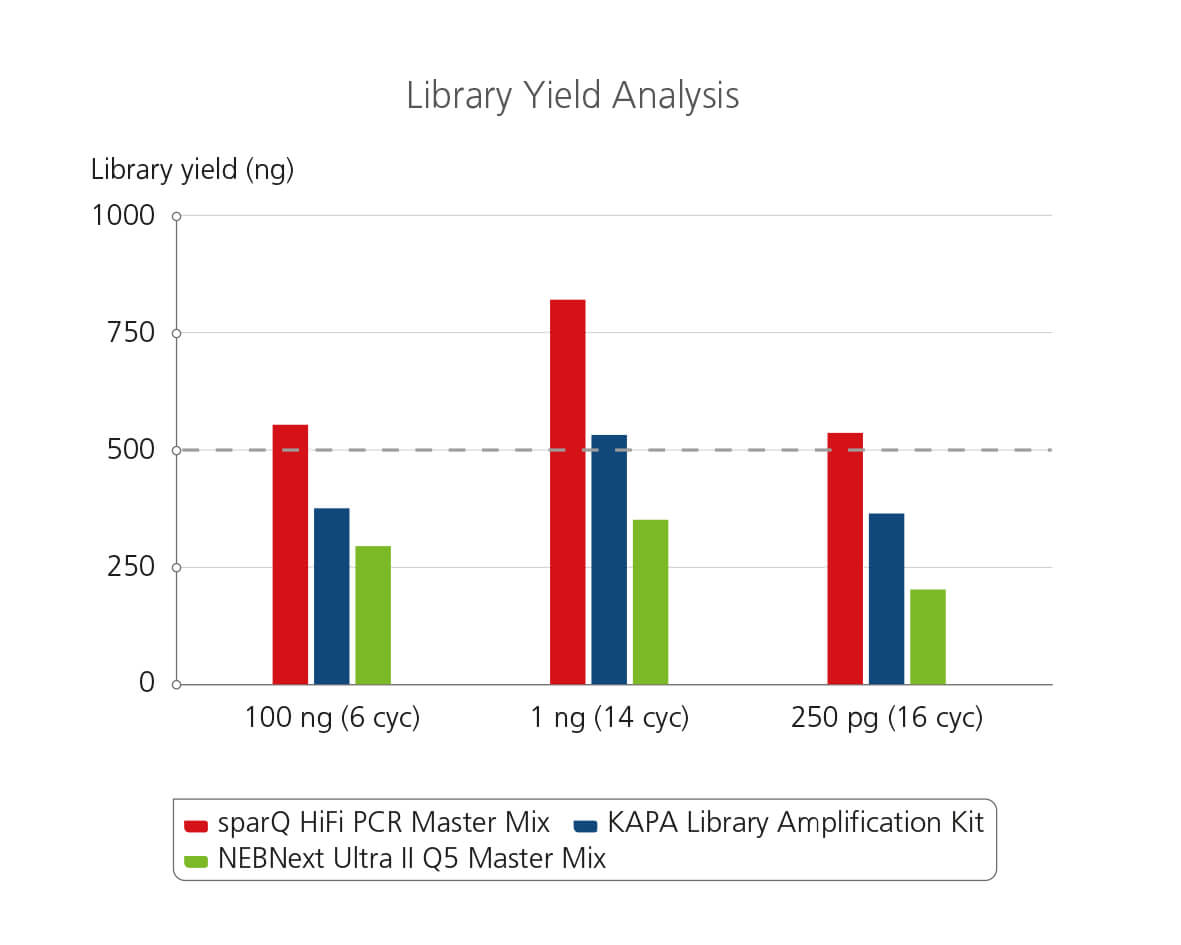
Libraries were prepared from Covaris-sheared DNA with sparQ DNA library prep kit prior to library amplification. Pre-amplified libraries were then amplified using sparQ HiFi PCR Master Mix (red), KAPA Library Amplification kit (blue), or NEBNext Ultra II Q5 Master Mix (green) with the identical amplification cycle numbers (6 cycles for 100 ng input DNA, 14 cycles for 1 ng input DNA, and 16 cycles for 250 pg input DNA). Amplified libraries were quantified with Qubit fluorometric quantitation method and qPCR-based quantification method (data not shown).
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews
sparQ HiFi PCR Master Mix
There are no reviews yet