
sparQ PureMag Beads
- High recovery of DNA fragments greater than 100 bp
- Efficient removal of unwanted components from adapter ligation and PCR reactions
- Consistent single or double-sided size selection
- Seamless integration into existing NGS workflows with little or no protocol change
- Easy-to-use and compatible with manual processing or automated liquid handling robots
- Cost effective alternative to AMPure® XP with equivalent performance
sparQ PureMag Beads
Description
sparQ PureMag Beads is a fast and reliable nucleic acid purification system for reaction cleanup and size selection in Next Generation Sequencing (NGS) workflows. Based on the reversible nucleic acid-binding properties of magnetic beads, this product can be used to quickly remove primers, primer-dimers, unincorporated nucleotides, salts, adapters and adapter-dimers from NGS library prep reactions to improve downstream sequencing performance. sparQ PureMag Beads allows excellent recovery of fragments greater than 100 bp without centrifugation or filtration. Consistent and reliable size selection can be achieved by simply adjusting the beads to sample ratio. This product is designed for both manual and automated processing, allowing seamless integration into existing workflows.

Beads performed as well in my tests as Ampure XP which I consider to be the gold standard. Impressive results given the lower price for sparQ PureMag Beads.”
Details
Performance Data
Efficient recovery of DNA equivalent to AMPure XP
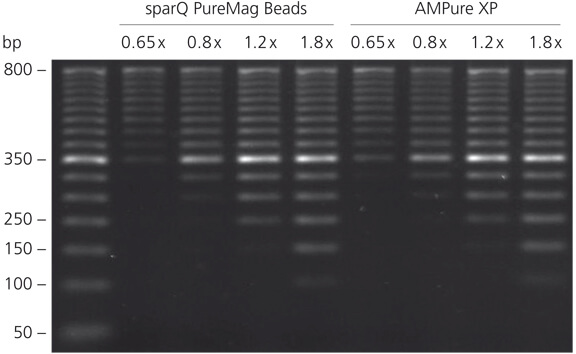
sparQ PureMag Beads show equivalent performance to AMPure XP for DNA purification. 50 bp DNA ladder was purified with sparQ PureMag Beads and AMPure XP at different beads to DNA ratios and analyzed on 2% agarose gel.
Highly reproducible purification across a range of inputs
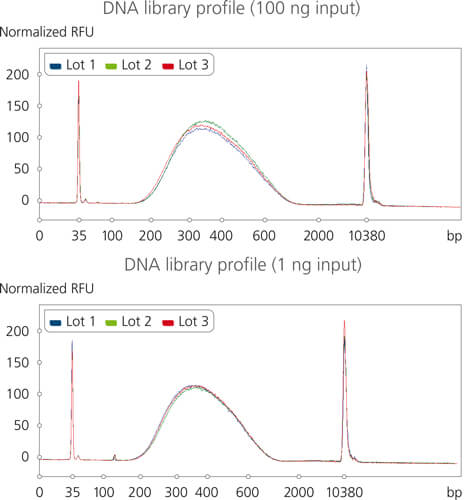
Highly reproducible DNA library profiles were achieved using different lots of sparQ PureMag Beads and a broad range of input amount. Libraries were prepared with sparQ DNA Library Prep Kit from 100 ng and 1 ng of fragmented microbial genomic DNA. sparQ PureMag Beads were used post adapter ligation and PCR amplification to effectively remove adapter-dimers and primer-dimers.
Bioanalyzer trace of fragmented human genomic DNA prior and post double-sided size selection
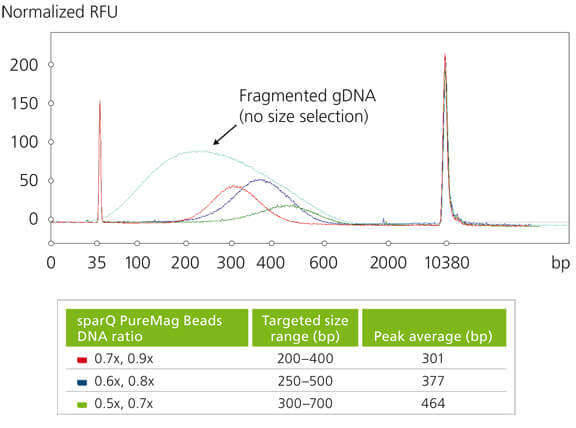
Electropherogram of fragmented human genomic DNA prior and post double-sided size selection. Different sparQ PureMag Beads to DNA ratios were used to achieve various targeted size range.
sparQ PureMag beads workflow
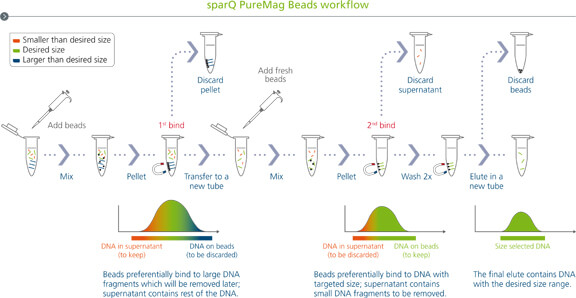
Double-sided size selection is used to remove smaller and larger fragments from either side of the desired region. The fragment size can be easily adjusted to suit the application by manipulating the sparQ PureMag Beads to DNA volumetric ratio.
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews
sparQ PureMag Beads
These beads performed similarly and followed a similar protocol to beads I have used in the past.
In head to head with beckman beads they performed the same. Not noticeable differences.
Equal performance and easy of use as our current beads.
They worked just fine. If the price is better, we can buy them!