sparQ DNA Frag & Library Prep Kit
- High quality libraries in 2.5 hours from 1 ng – 1 μg of input DNA
- Tunable and reproducible fragmentation size range
- Simple, convenient 2-step workflow with minimal hands-on time
- Novel chemistry and high-fidelity amplification minimizing bias
- Superior sequence coverage uniformity and low duplication rate
sparQ DNA Frag & Library Prep Kit is intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.
sparQ DNA Frag & Library Prep Kit
Description
The sparQ DNA Frag & Library Prep Kit is optimized for enzymatic fragmentation of DNA and streamlined construction of high quality libraries for sequencing on Illumina NGS® platforms. The simple, convenient 2-step workflow can be completed in 2.5 hours with minimal hands-on time and accommodates DNA input amounts from 1 ng to 1000 ng. The DNA fragmentation and polishing reactions are combined in a single step to generate 5'-phosphorylated and 3'-dA-tailed fragments. This is followed by high efficiency adapter ligation in the same tube. PCR-Free workflows are enabled from 100 ng of starting material. If library amplification is required, the HiFi PCR Master Mix and Primer Mix ensure even amplification with minimal bias.
NGS Automation
Quantabio sparQ Frag & Library Prep Kit can be fully automated
Currently sparQ Frag & Library Prep Kit is supported by the following vendors:
| Vendor | Automation Platform(s) |
| PerkinElmer Applied Genomics | SCICLONE G3 NGSx |
| Tecan | DreamPrep® NGS |
| Beckman Coulter | in development |
Looking for more information or for different automation vendor?
sparQ UDI Adapter Resources
Setting up sparQ Library Prep kits in Illumina Experiment Manager
Watch the step-by-step tutorial illustrating the sparQ DNA Frag & Library Prep Kit protocol
sparQ NGS Library Prep Solutions

The sparQ kit safeguards samples from over fragmentation and routinely provides more consistent fragment size. Once optimized, I had peace of mind knowing my samples were safe when I got busy in the lab.
Details
|
|
|
Volume |
|
|
Component Description |
Cap Color |
24 Reactions |
96 Reactions |
|
DNA Frag & Polishing Enzyme Mix (5X) |
Blue |
1 x 240 µl |
1 x 960 µl |
|
DNA Frag & Polishing Buffer (10X) |
Blue |
1 x 120 µl |
1 x 528 µl |
|
DNA Frag & Polishing Enhancer Solution |
Blue |
1 x 288 µl |
1 x 264 µl |
|
DNA Ligase |
Orange |
1 x 240 µl |
1 x 960 µl |
|
DNA Rapid Ligation Buffer (5X) |
Orange |
1 x 480 µl |
2 x 960 µl |
|
HiFi PCR Master Mix (2X) |
White |
1 x 600 µl |
2 x 1.2 ml |
|
Primer Mix |
White |
1 x 72 µL |
1 x 144 µl |
Performance Data
Quality Sequencing metrics and improved workflow economics
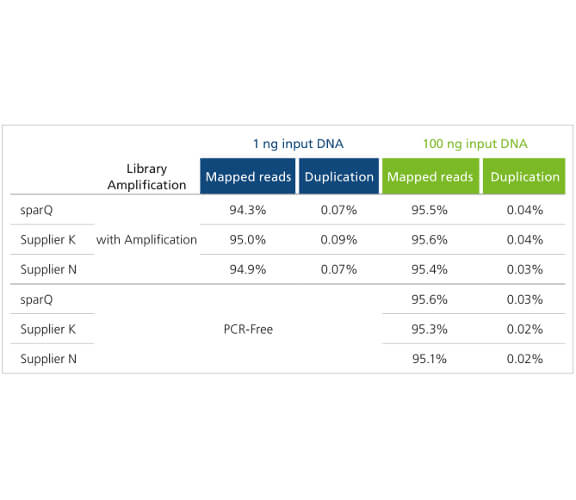
sparQ DNA Frag & Library Prep Kit generates high quality DNA libraries with minimal duplication artifacts. Libraries were prepared with 1 ng and 100 ng of microbial genomic DNA, amplified for 12 and 6 cycles respectively, and subsequently sequenced on Illumina MiSeq. Each library was down-sampled to 2 million reads (150 bp paired-end reads) and aligned to a reference genome with only unique alignments reported.
Genome coverage analysis
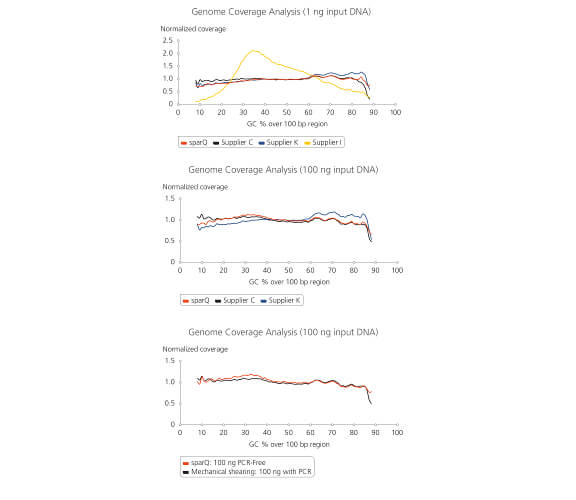
Library prepared using sparQ DNA Frag & Library Prep Kit resulted in uniform coverage across the wide range of GC-content. Libraries were prepared using different DNA fragmentation and library preparation kits with 1 ng or 100 ng of microbial gDNA followed by sequencing on Illumina MiSeq. 2 million reads from each tested library were down-sampled and analyzed. Coverage uniformity against GC-content bias resulting from different DNA fragmentation and library preparation kits were compared by plotting normalized coverage for a wide GC-content.
Libraries prepared using PCR-free workflow of sparQ DNA Frag & Library Prep Kit with 50 ng of microbial genomic DNA shows similar high performance as a typical amplified library prepared by Covaris mechanical shearing method.
Yield comparison
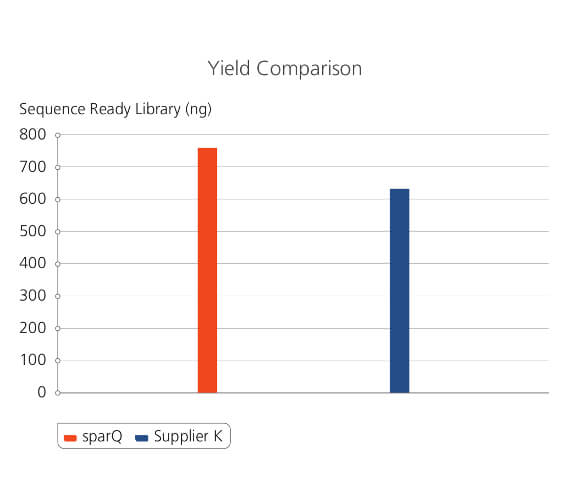
sparQ DNA Frag & Library Prep Kit shows significantly higher NGS library preparation efficiency. Libraries with 300 bp average DNA fragments from 100 ng of gDNA Coriell NA12878 were prepared using sparQ DNA Frag & Library Prep Kit and a commercial kit. Manufactures’ manuals were carefully followed. Amplified libraries (5 cycles of amplification) were quantified by Qubit fluorometric quantitation method
Fragmentation tuning guide
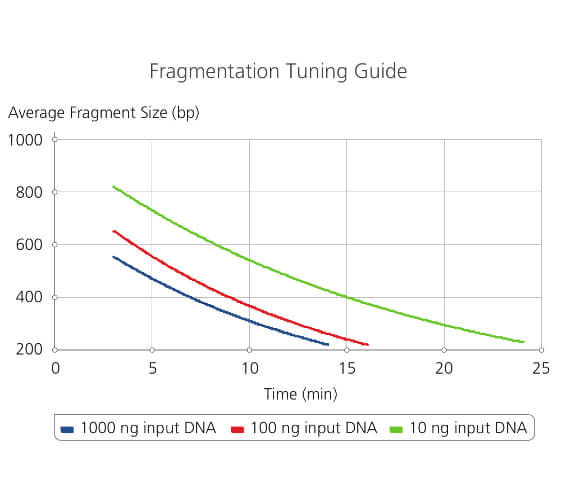
Simply select the desired fragment size and input DNA amount. If input DNA falls between values displayed on the graph, an estimate can be used for optimizing fragmentation times.
Streamlined workflow
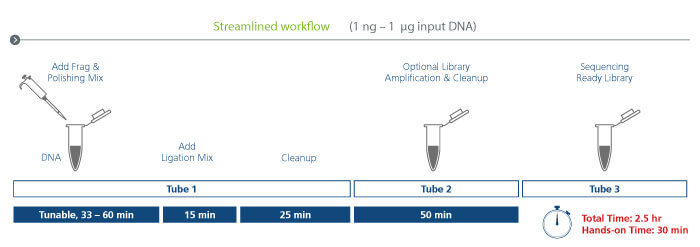
The streamlined workflow utilizes a proprietary enzyme mix that integrates tunable and reproducible fragmentation with DNA polishing simplifying library construction. The same single reaction tube is used to proceed to adapter ligation and cleanup, minimizing sample transfer steps. A second tube is used for workflows requiring PCR amplification, and a final tube receives the sequencing-ready library.
Fragmentation time course
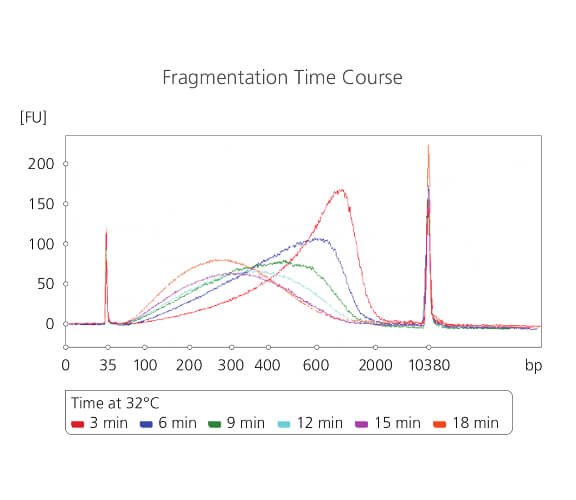
sparQ DNA Frag & Library Prep Kit is tunable to the desired fragment size. 100 ng Human gDNA was subjected to fragmentation with a series of incubation time points (3 – 18 min). After fragmentation, DNA samples were purified and then visualized using Agilent High Sensitivity DNA Kit.
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews
| 5 star | 75% | |
| 4 star | 25% | |
| 3 star | 0% | |
| 2 star | 0% | |
| 1 star | 0% |
sparQ DNA Frag & Library Prep Kit
Customer Testimonials
Welcome to the Quantabio webshop!
Please complete the new user account registration form
Once your account is setup, you'll be able to purchase any of the products on our site at any time with next day delivery for in stock products.
I do not have a lot of experience with other similar products for comparison, but overall this kit was great and the protocol was very clear. I was working with samples of degraded or low quality DNA, but still managed to get usable data from most of the samples.
I have had the opportunity to work with the sparQ DNA Frag & Library Prep Kit twice at the time of this survey. Overall, the kit has performed well with results that are comparable in quality to those obtained using the standard Illumina DNA Prep Kit. During the second test run, a second analyst also ran samples that were processed by the same kit. Unfortunately, some of metrics for this analyst’s samples fell just below the established FDA Genometrakr standards for NCBI submission. I feel that this is an anomaly, and I cannot confidently say that it is the result of any issues with this kit. We are currently in the process of validating the kit to show that it has the potential to be a valuable alternative means for DNA library processing for WGS. I am convinced that with additional use and increased familiarity with the procedure, we will be able to further fine tune the protocol to achieve consistently high-quality data. Overall, the experience has been positive, and the major benefits include the ease of use, reduced processing time, and a lower risk of inadvertent loss of sample DNA thanks to fewer steps that involve the transfer of supernatant from well to well.
There should be a small kit to convert “sparQ DNA Library Prep Kit 96R” into “sparQ DNA Frag & Library Prep Kit 96R”. It is only the matter of adding couple of enzymes and buffers for fragmentation.
The library prep was a little longer than our current kit, and the specifications for PCR lid and block temperature prior to PCR start and to work on ice made the procedure more tedious than our current workflow. The library output was generally a little higher than our current kit (with the same amount of PCR cycles). The sequencing output was similar to our current kit.