sparQ DNA Library Prep Kit
- Fast, easy single-tube solution completes library prep in 2.5 hours
- Suitable for a wide range of input amounts from as low as 250 pg
- Optimized chemistry ensuring superior library prep sensitivity and efficiency
- Higher library yields compared to other library prep kits
- High efficiency enables PCR-free workflow from 100 ng input
sparQ DNA Library Prep Kit is intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.
sparQ DNA Library Prep Kit
Description
sparQ DNA Library Prep Kit is optimized for the rapid construction of DNA libraries from fragmented double-stranded DNA for sequencing on Illumina® NGS platforms. The simplified protocol speeds up library prep to 2.5 hours with minimal hands-on time and accommodates DNA input amounts from 250 pg to 1 ug. DNA polishing reactions are streamlined into a single step to convert fragmented DNA into 5'-phosphorylated and 3'-dA-tailed DNA fragments. This is followed by high efficiency adapter ligation in the same tube. PCR-free workflows are enabled from 100 ng of starting material. If library amplification is required, the HiFi PCR Master Mix and Primer Mix ensure even amplification with minimal bias.
NGS Automation
Quantabio sparQ Frag & Library Prep Kit can be fully automated
Currently sparQ Frag & Library Prep Kit is supported by the following vendors:
| Vendor | Automation Platform(s) |
| PerkinElmer Applied Genomics | SCICLONE G3 NGSx |
| Tecan | DreamPrep NGS |
| Beckman Coulter | in development |
Looking for more information or for different automation vendor?
sparQ UDI Adapter Resources
Setting up sparQ Library Prep kits in Illumina Experiment Manager
Watch the step-by-step tutorial illustrating the sparQ DNA Library Prep Kit protocol
sparQ NGS Library Prep Solutions

Details
|
|
|
Volume |
|
|
Component Description |
Cap Color |
95191-024 |
95191-096 |
|
DNA Polishing Enzyme Mix |
Blue |
1 x 0.24 ml |
1 x 0.96 ml |
|
DNA Polishing Buffer |
Blue |
1 x 0.12 ml |
1 x 0.48 ml |
|
DNA Ligase |
Orange |
1 x 0.24 ml |
1 x 0.96 ml |
|
DNA Rapid Ligation Buffer |
Orange |
1 x 0.48 ml |
2 x 0.96 ml |
|
HiFi PCR Master Mix (2X) |
White |
1 x 0.60 ml |
2 x 1.25 ml |
|
Primer Mix |
White |
1 x 0.072 ml |
1 x 0.144 ml |
Performance Data
sparQ DNA Library Prep Kit Yield Analysis
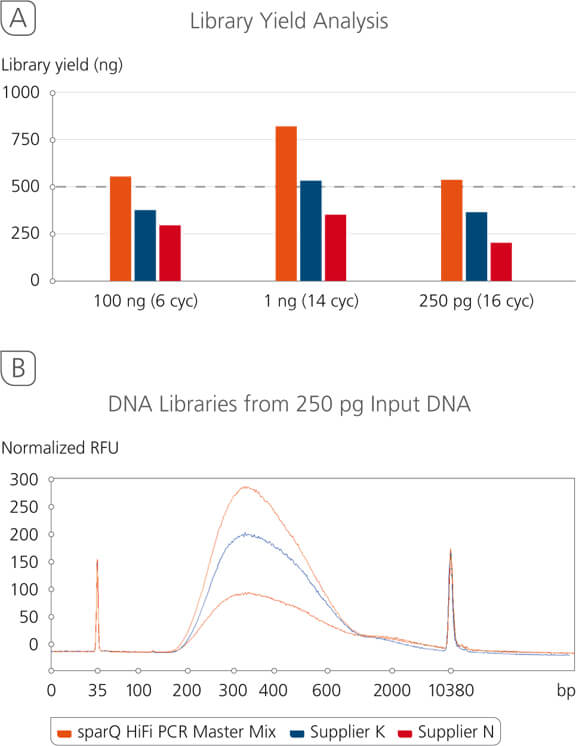
sparQ DNA Library Prep Kit produces high quality libraries from a broad range of DNA inputs with significantly higher yields. Libraries were prepared with Covaris-sheared DNA (250 bp average size) using kit manufacturers’ instructions. Amplified libraries (6 amplification cycles for 100 ng input DNA and 13 amplification cycles for 1 ng input DNA) were quantified with Qubit fluorometric quantitation method.
Improve Sequencing Results and Economics
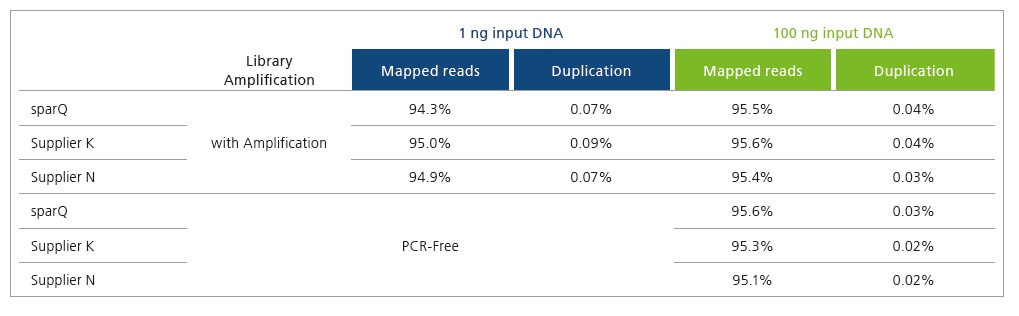
sparQ DNA Library Prep Kit generates high quality DNA libraries with minimal duplication rates. Libraries were prepared with 1 ng and 100 ng of microbial genomic DNA and subsequently sequenced on Illumina MiSeq. Each library was down-sampled to 2 million reads (150 bp paired-end reads) and aligned to a reference genome with only unique alignments reported.
Uniform coverage across wide range of GC-content
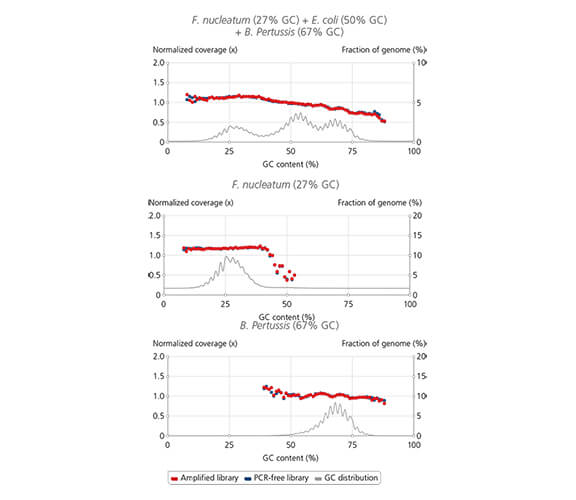
Library amplification with sparQ HiFi PCR Master Mix contained in the sparQ DNA Library Prep Kit resulted in uniform coverage across the wide range of GC-content. Libraries were prepared by using sparQ DNA Library Prep Kit with 100 ng input DNA. Coverage depth against GC-content of libraries amplified by sparQ HiFi PCR Master Mix (orange) were compared to corresponding libraries without amplification (dark blue: PCR-free library). GC content distribution of targeted genomes is indicated by gray line.
sparQ DNA Library Prep Workflow

The streamlined workflow can be completed in under 3 hours with minimal hands-on time. A single tube is used for DNA polishing, ligation, and cleanup. A second tube is used for workflows requiring PCR amplification and a final tube receives the sequencing-ready library.
Coverage Across Challenging Regions
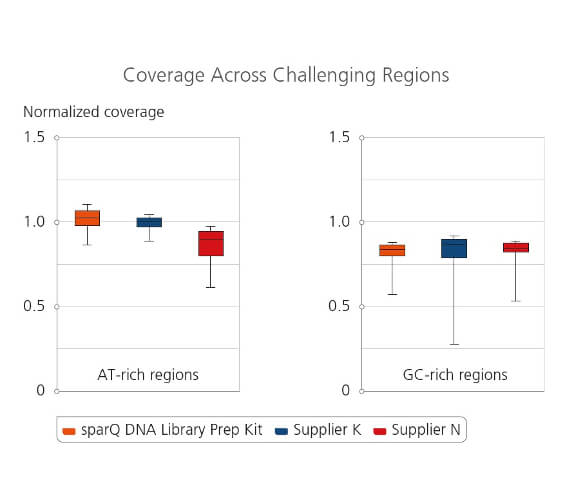
Amplified libraries were prepared from 100 ng of microbial genomic DNA and subsequently sequenced on Illumina MiSeq. 2 million reads from each tested library were down-sampled and analyzed. Coverage uniformity for different library preparation kits were compared by plotting normalized coverage for both extreme AT-rich regions (8%-20% GC-content) and GC-rich regions (75%-88% GC-content).
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews
sparQ DNA Library Prep Kit
LP worked great – it was fast and easy. The minimum DNA input was much lower than our current kit which would allow us to recover samples that previously would fail. Definitely something we’ll look into using in the next pipeline update. Thanks!