PerfeCTa MultiPlex qPCR SuperMix
- Maximum assay sensitivity and target precision with stringent, ultrapure, and high-yielding AccuStart II hot start Taq DNA polymerase
- Broad linear detection range with cDNA or genomic DNA templates
- Enables highly sensitive 4 target detection assays with coamplified, robust internal positive controls
- Supports efficient vortex mixing with proprietary anti-foaming technology
PerfeCTa Multiplex qPCR SuperMix is intended for molecular biology applications. This product is not intended for the diagnosis, prevention or treatment of a disease.
PerfeCTa MultiPlex qPCR SuperMix Low ROX
PerfeCTa MultiPlex qPCR SuperMix
Description
PerfeCTa Multiplex qPCR SuperMix is a 2X concentrated, ready-to-use reaction cocktail that contains all the necessary components except: primers, probe(s), and DNA template for highly-multiplexed, real-time quantitative PCR. This reagent formulation pushes the boundary of multiplex qPCR by enabling unbiased amplification of up to 5 targets in a single amplification reaction. Suppression of low abundance targets by high abundance reference targets during co-amplification is a common problem in multiplex PCR in which individual assay sensitivity can be significantly compromised. PerfeCTa Multiplex qPCR SuperMix, Low ROX delivers assay performance with exceptionally broad, linear detection and limit-of-detection (LOD) sensitivity to multiplexed qPCR that is comparable single-plex assay performance without the need for rigorous titration of individual primer assays. A key component of this SuperMix is ultra-pure AccuStart hot start Taq DNA polymerase that is completely arrested prior to the initial PCR denaturation step. Upon heat activation at 95°C, the antibodies are rapidly and irreversibly denatured, releasing a fully active high-yielding Taq DNA polymerase mutant. This enables specific and efficient primer extension with the convenience of ambient room-temperature reaction assembly.
Details
2X concentrated SuperMix Reagent, containing:
- Reaction buffer with multiplex qPCR-optimized MgCl2, dATP, dCTP, dGTP, and dTTP.
- Ultra-pure AccuStart hot start Taq DNA Polymerase.
- Reference dye (if applicable).
- Proprietary performance enhancing additives and stabilizers.
Instrument Compatibility
ROX
- Applied Biosystems 5700
- Applied Biosystems 7000
- Applied Biosystems 7300
- Applied Biosystems 7700
- Applied Biosystems 7900
- Applied Biosystems 7900HT
- Applied Biosystems 7900 HT Fast
- Applied Biosystems StepOne™
- Applied Biosystems StepOnePlus™
Low ROX
- Applied Biosystems 7500
- Applied Biosystems 7500 Fast
- Stratagene Mx3000P®
- Stratagene Mx3005P™
- Stratagene Mx4000™
- Applied Biosystems ViiA 7
- Applied Biosystems QuantStudio™ (all models)
- Douglas Scientific IntelliQube®
No ROX
- Quantabio Q
- BioRad CFX
- Roche LightCycler 480
- QIAGEN Rotor-Gene Q
- Agilent AriaMx
- Azure Cielo™
- Analytik Jena qTower
- Analytik Jena qTOWERiris
- Other
Bio-Rad iCycler iQ systems
- BioRad iCycler iQ™
- BioRad MyiQ™
- BioRad iQ™5
Performance Data
Four-color multiplex comparison: extended cycling conditions.
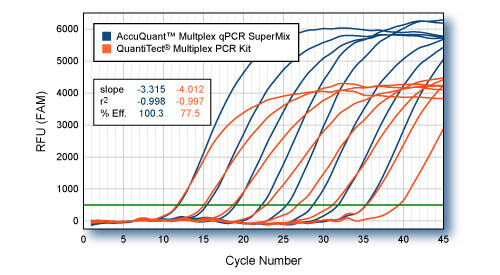
Varying amounts of GAPD plasmid DNA (10 to 1 x 107 copies) were co-amplified with 1 x 108 copies (each) of beta-actin (ACTB), interleukin-1beta (IL1B), and α-tubulin (TUB). 4-color multiplexed qPCRs were carried out in 50-μl volumes with PerfeCTa™ Multiplex qPCR SuperMix or QuantiTect® Multiplex PCR Kit (Qiagen) according to each manufacturers recommend conditions. Reactions were cycled according to the Qiagen recommended protocol: 15 min, 95°C, followed by 45 cycles of 1 min, 94°C, 1.5 min, 60°C. Average of four replicate reactions for each input amount of GAPD plasmid (FAM-labeled probe) are shown. qPCR of ACTB (HEX-labeled probe), IL1B (Texas Red-Labeled probe), and TUB (Cy5-labelled probe) not shown. All probes utilized non-fluorescent Black Hole Quencher® (BioSearch Technologies). Suppression of GAPD Cts are evident in reactions with the QuantiTect reagents when competing amplicons are >1000-fold excess over the limiting target sequence (GAPD). PerfeCTa™ Multiplex qPCR SuperMix accurately amplifies GAPD target in the presences of >1 x 107-fold excess of competing amplicons.
Four-color multiplex comparison: fast cycling conditions
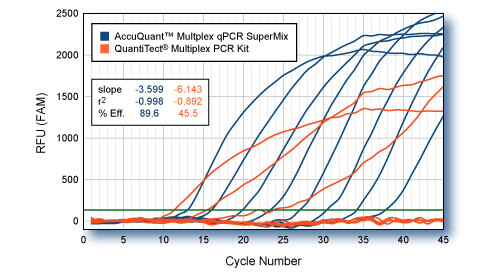
Varying amounts of GAPD plasmid DNA (10 to 1 x 107 copies) were co-amplified with 1 x 108 copies (each) of beta-actin (ACTB), interleukin-1beta (IL1B), and α-tubulin (TUB). 4-color multiplexed qPCRs were carried out in 25-μl volumes with PerfeCTa™ Multiplex qPCR SuperMix or QuantiTect® Multiplex PCR Kit (Qiagen). Primer and probe concentrations were according to each manufacturers recommend conditions. Each hot-start Taq was activated according to conditions specified for each product. QuantiTect reagents were incubated for 15 min at 95C, where as PerfeCTa Multiplex reactions were incubated for 3 min at 95C. All reactions were cycled for 45 cycles of 10s, 95°C, 30s, 60°C. Average of four replicate reactions for each input amount of GAPD plasmid (FAM-labeled probe) are shown. qPCR of ACTB (HEX-labeled probe), IL1B (Texas Red-Labeled probe), and TUB (Cy5-labelled probe) not shown. All probes utilized non-fluorescent Black Hole Quencher® (BioSearch Technologies). Significant degradation of performance with the QuantiTect reagents using abbreviated cycle times is readily apparent. However, recovery of full and unmodified Taq DNA polymerase activity afforded by PerfeCTa™ Multiplex qPCR SuperMix enables highly efficient multiplex qPCR even with rapid cycling conditions.
Documents & Downloads
 Powered by Bioz
Powered by Bioz
Customer Product Reviews
PerfeCTa MultiPlex qPCR SuperMix
There are no reviews yet