

qScript Ultra SuperMix

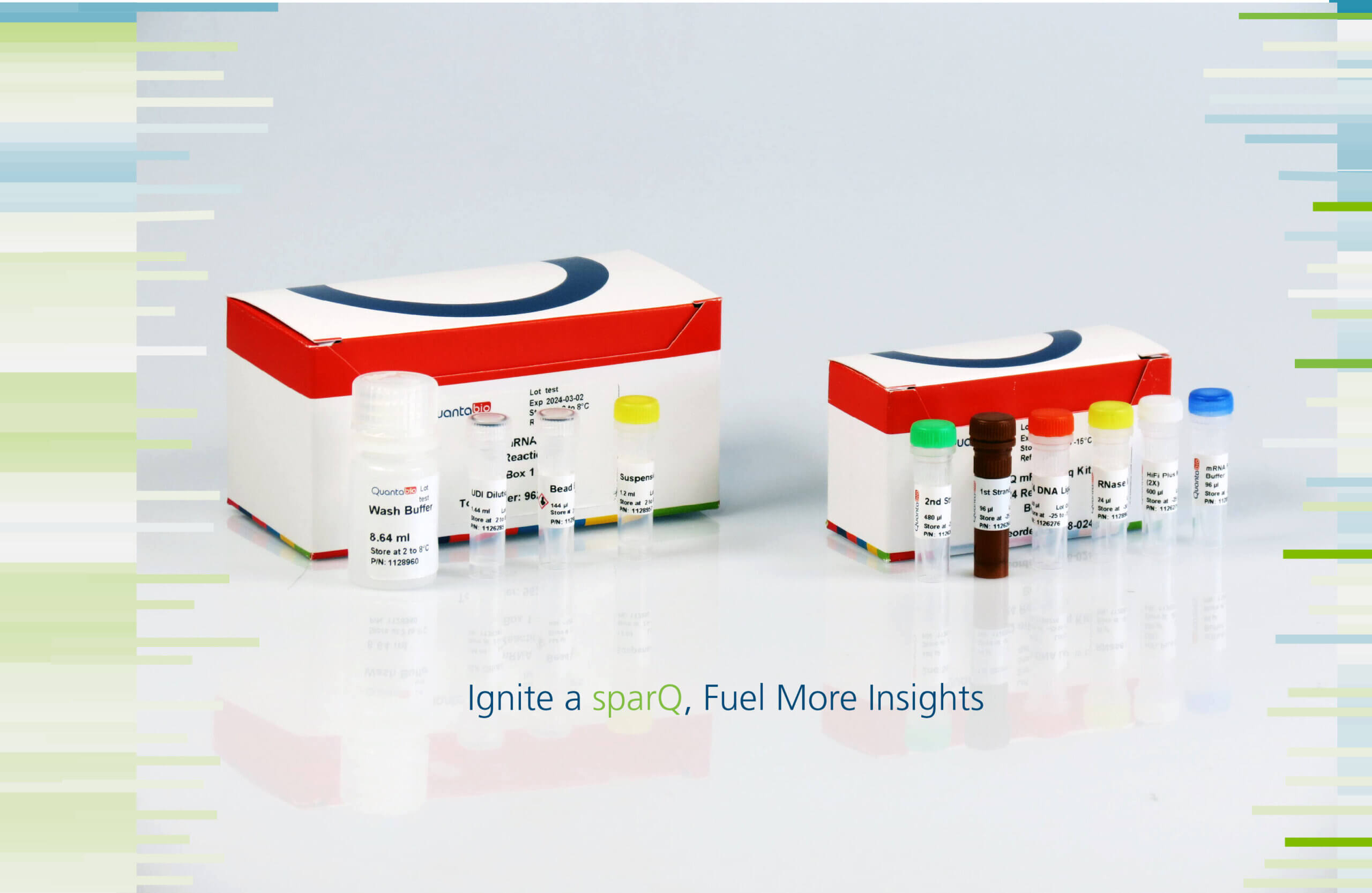
sparQ mRNA-Seq Kit

Explore repliQa HiFi ToughMix
Enable Better Library Amplification For a Range of NGS Applications

16S Metagenomic Sequencing
Rapid 16S Metagenomic Library Preparation for Oxford Nanopore Technologies (ONT)® Platform

GC-Rich Template Amplification
Significant improvements to PCR amplification enables better performance for a multitude of Next Generation Sequencing applications

Short and Long Read Sequencing
Identifying the best PCR enzyme for library amplification in NGS for both short and long read sequencing

Avoid DNA polymerase, nucleic acid, and other common PCR inhibitors in crude blood samples with Quantabio ToughMixes.
Overcome the limitations of sensitivity and specificity in dried blood samples with Quantabio’s advanced ToughMixes.

Our ToughMix reagents withstand common PCR-suppressive compounds in plant & soil samples, such as phenols, polysaccharides, and humic acid.
& Reduce Costs
Quantabio offers complete library prep solutions for Illumina NGS platforms. High-quality reagents deliver unmatched efficiency and robust performance to ensure reliable and reproducible sequencing results while reducing total costs.


Size Selection

From product brochures and data sheets to trade publications and webinars, we have the most up-to-date information you’re looking for here.